DOI:https://doi.org/10.47990/alop.v12i1.367
Lelimar Rosbelt Palencia Torres1, Josnelly Betania Santaella Pantoja1, Ysabel Cristina Zamudio Acosta2, Neorlay Desireé Rondón Jiménez3
Resumen: El Síndrome de Cornelia de Lange (SCDL), es una anomalía genética cuya prevalencia es de 1:62.000- 1:45.000 de los nacimientos. Se atribuye principalmente a mutaciones en los genes NIPBL, SMC3 y SMC1A. Se caracteriza por presentar alteraciones físicas generales, alteración cognitiva y del lenguaje; y rasgos orofaciales como la sinofridia, hirsutismo, también existe maloclusión, retardo de la erupción, apiñamiento, anodoncia, malformación de las extremidades, retraso del desarrollo pre y postnatal y otras malformaciones congénitas. Objetivo: Analizar el caso de paciente con síndrome de Cornelia de Lange y su relación con algunos hallazgos reportados en la literatura especialmente la erupción dentaria. Se presenta paciente lactante femenina de 2 años y 5 meses, procedente de Valencia, con diagnóstico genético de Síndrome de Cornelia de Lange, plumbemia, y litiasis biliar, que acude a la consulta del Postgrado de Odontopediatría de la Universidad de Carabobo por presentar retardo en la erupción dentaria. Se realiza historia clínica, examen clínico general donde se observa retraso psicomotor, del lenguaje y características fenotípicas propias del síndrome. A la evaluación clínica intrabucal se observa rebordes gingivales con inserción normal de frenillos y ausencia de unidades dentarias (retardo de erupción). La erupción dentaria puede verse afectada en pacientes con diagnóstico de Síndrome de Cornelia de Lange, tanto en su cronología como en la secuencia de erupción.
Palabras clave: Síndrome de Cornelia de Lange, retardo de la erupción, secuencia de erupción.
Resumo: A síndrome de Cornelia de Lange (SCDL) é uma anormalidade genética com prevalência de 1: 62.000-1: 45.000 de nascimentos. É atribuído principalmente a mutações nos genes NIPBL, SMC3 e SMC1A. Caracteriza-se por apresentar alterações físicas gerais, alteração cognitiva e de linguagem; e características orofaciais, como sinofrídios, hirsutismo, também há má oclusão, erupção retardada, aglomeração, anodontia, malformação de membros, atraso no desenvolvimento pré-natal e pós-natal e outras malformações congênitas. Objetivo: Analisar o caso de um paciente com síndrome de Cornelia de Lange e sua relação com alguns achados relatados na literatura, principalmente erupção dentária. Apresentamos uma paciente de enfermagem de Valência com 2 anos e 5 meses de idade, com diagnóstico genético da síndrome de Cornelia de Lange, plumbemia e litíase biliar, que compareceu à consulta de pós-graduação em Odontopediatria da Universidade de Carabobo por apresentar atraso erupção dentária. História clínica, exame clínico geral, onde são observados retardo psicomotor, linguagem e características fenotípicas da síndrome. Uma avaliação clínica intraoral mostra sulcos gengivais com inserção normal de aparelho e ausência de unidades dentárias (erupção tardia). A erupção dentária pode ser afetada em pacientes diagnosticados com Síndrome de Cornelia de Lange, tanto na cronologia quanto na sequência da erupção.
Palabra-chave: síndrome de Cornelia de Lange, atraso na erupção, sequência de erupção.
Abstract: Cornelia de Lange Syndrome (SCDL) is a genetic abnormality with a prevalence of 1: 62,000- 1: 45,000 of births. It is mainly attributed to mutations in the NIPBL, SMC3 and SMC1A genes. It is characterized by presenting general physical alterations, cognitive and language alteration; and orofacial features such as sinofridia, hirsutism, there is also malocclusion, delayed eruption, crowding, anodontia, limb malformation, prenatal and postnatal developmental delay, and other congenital malformations. Objective: To analyze the case of a patient with Cornelia de Lange syndrome and its relationship with some findings reported in the literature, especially dental eruption. We present a 2-year-old and 5-month-old female nursing patient from Valencia with a genetic diagnosis of Cornelia de Lange Syndrome, plumbemia, and biliary lithiasis, who attended the Pediatric Dentistry Postgraduate consultation at the University of Carabobo for presenting delay in tooth eruption. Clinical history, general clinical examination where psychomotor retardation, language and phenotypic characteristics of the syndrome are observed. A clinical intraoral evaluation shows gingival ridges with normal insertion of braces and absence of dental units (delayed eruption). The dental eruption can be affected in patients diagnosed with Cornelia de Lange Syndrome, both in its chronology and in the eruption sequence.
Key words: Cornelia de Lange Syndrome, Tooth eruption delay, eruption sequence.
El Síndrome de Cornelia de Lange (SdCL) es una anomalía genética caracterizada por presentar un fenotipo facial distintivo en el que se destacan rasgos craneofaciales peculiares, malformación de las extremidades, retraso del desarrollo pre y postnatal, afectación a nivel cognitivo, particularmente del lenguaje, y otras malformaciones congénitas1,2. Fue descrito por primera vez en el año 1933 por la Dra. Cornelia de Lange en dos niñas3. La prevalencia es variable según los estudios publicados, oscilando entre 1:62.000- 1:45.000 nacimientos4. Aunque la mayoría de los casos son esporádicos, existen casos familiares con un patrón de herencia dominante, incluyendo casos con mosaicismo germinal. Se han determinado tres genes causales: NIPBL, SMC1A y SMC3 que codifican proteínas reguladoras o estructurales del Complejo de Cohesinas5,6. En el año 2004 se describió el primer gen asociado al SdCL, denominado NIPBL y posteriormente se identificaron dos genes más el SMC1A y el SMC37,8.
Este síndrome se produce por afectación de los genes que codifican proteínas reguladoras o estructurales del complejo de cohesinas. La cardiopatía congénita (CC) no es criterio mayor de enfermedad, pero afecta a numerosos individuos. En un estudio realizado en España en el año 2017; donde se evaluaron 149 niños con SdCL se demostró que un 34,9% de los pacientes presentó algún tipo de cardiopatía congénita9.
Clínicamente se distinguen tres fenotipos: leve, moderado y grave. Leve, sin defecto de reducción de las extremidades; moderado, defecto de reducción parcial/oligodactilia menor a 2 dígitos en cualquier mano; y grave, defecto de reducción de las extremidades grave/oligodactilia mayor a 2 dígitos en cada mano. Los parámetros de crecimiento: leve, parámetros de crecimiento promedio y mayor a percentil 75 en las curvas de crecimiento SdCL; moderada, parámetros de crecimiento promedio y percentil 25- 75 en las curvas de crecimiento SdCL; y grave, parámetros de crecimiento promedio y menor a percentil 25 en las curvas de crecimiento SdCL. En cuanto a la función cognitiva el fenotipo leve mayor a 2 años de retraso de los estándares normales, con el desarrollo de las habilidades del habla y de la comunicación en las personas de edad avanzada; moderado, > 2 años atrás de los estándares de desarrollo normales, con habla y comunicación limitada; grave, retraso profundo, con una falta de comunicación significativa10,11 (Tabla 1).
En ese mismo sentido se muestra retraso de crecimiento intrauterino y postnatal, que se ha registrado en tablas de crecimiento específicas para este síndrome; por ejemplo, en neonatos se ha reportado el peso, talla y perímetro cefálico en el percentil menos 3. Asimismo, estos recién nacidos presentan problemas de alimentación los primeros meses o años de vida por la presencia de anormalidades gastrointestinales (muchos necesitan sonda nasogástrica) que contribuyen a la persistencia de este problema 7.
En muchos pacientes con SdCL se evidencia trastorno del espectro autista (ASD), comportamiento autolesivo, agresivo e hiperactividad12-17. Además presentan manifestaciones bucales como: micrognatia, apiñamiento dental, enfermedad periodontal, hipoplasia del esmalte, erosión de esmalte y dentina ocasionada por reflujo gastroesofágico, atresia de los arcos dentales y retraso en la erupción dental 18-20.
El proceso de erupción dentaria está genéticamente asociado con genes como el POST, RUNX2, AMELX. Además, es bien sabido que la cavidad bucal forma parte del sistema estomatognático y, por lo tanto; al estar alterado alguno de sus componentes se desencadenan una serie de eventos que afectan dicho sistema. Y al presentarse un retraso en el crecimiento y en la erupción dentaria, se va a presentar un desarrollo reducido del maxilar y la mandíbula por la poca estimulación neuromuscular, lo que nos va a traer como consecuencia que el paciente presente algún tipo de maloclusión. Y donde el tiempo de erupción va a depender de la herencia, el metabolismo óseo, el periodo de desarrollo y la posición fetal, las hormonas, la raza, la nutrición, enfermedades que ha sufrido el individuo y una diversidad de factores locales21.
Tabla 1: Características del Síndrome Cornelia de Lange
Se han documentado diversos factores etiológicos asociados al retraso de la erupción como dientes supernumerarios, anquilosis, quistes, erupción ectópica, tumores odontogénicos o no odontogénicos y deficiencias nutricionales. Por lo tanto, los genes que se relacionan con el desarrollo craneofacial tienen alguna injerencia sobre el proceso eruptivo y estos a su vez con variados factores epigenéticos22-24.
El síndrome de Cornelia de Lange es muy poco común de ver y está asociado a múltiples afecciones generales y bucales que nos conciernen especialmente a quienes nos dedicamos a la odontopediatria. Es importante conocer estas afecciones y saber relacionarlas con el síndrome a fin de orientar y guiar a los padres y al paciente para que éste tenga un mejor desarrollo y mejor calidad de vida. El objetivo de este artículo es analizar el caso de paciente con síndrome de Cornelia de Lange y su relación con algunos hallazgos reportados en la literatura.
Paciente lactante femenina de 2 años y 5 meses, procedente y natural de Valencia Edo. Carabobo, Venezuela, con diagnóstico genético de Síndrome de Cornelia de Lange moderado. Acude a la consulta acompañada de su madre, al Postgrado de Odontopediatría de la Universidad de Carabobo por preocupación por la ausencia de unidades dentarias. Principalmente se inició con la autorización de la madre, firmando un consentimiento informado, y cumpliendo con las declaraciones de Helsinki25. Aunque para este caso, utilizamos una referencia internacional como lo es la declaración de Helsinki; en Venezuela existe el código de ética para la vida que fue creado durante el gobierno actual de este País 26.
A la anamnesis madre refiere que su hija presenta, hipercalciuria, estenosis valvular pulmonar leve, reflujo gastroesofágico y litiasis biliar, también bajo peso (3,300 kg) y talla (46 cm) al nacer. Su informe médico refiere que en noviembre de 2015 le evidenciaron altos niveles de plomo en sangre: 7 μg/dl (VN: Niños: <5 μg/dl. Técnica: NIOSH). Sin embargo, no hay antecedentes de exposición al Pb: ni ambientales, ni laborales; aun cuando, expresan cuestionamientos acerca de la calidad del agua que reciben. Finalmente, control hematológico revela serie roja normocítica. Serie blanca leucocitosis. Plaquetas: morfología normal.
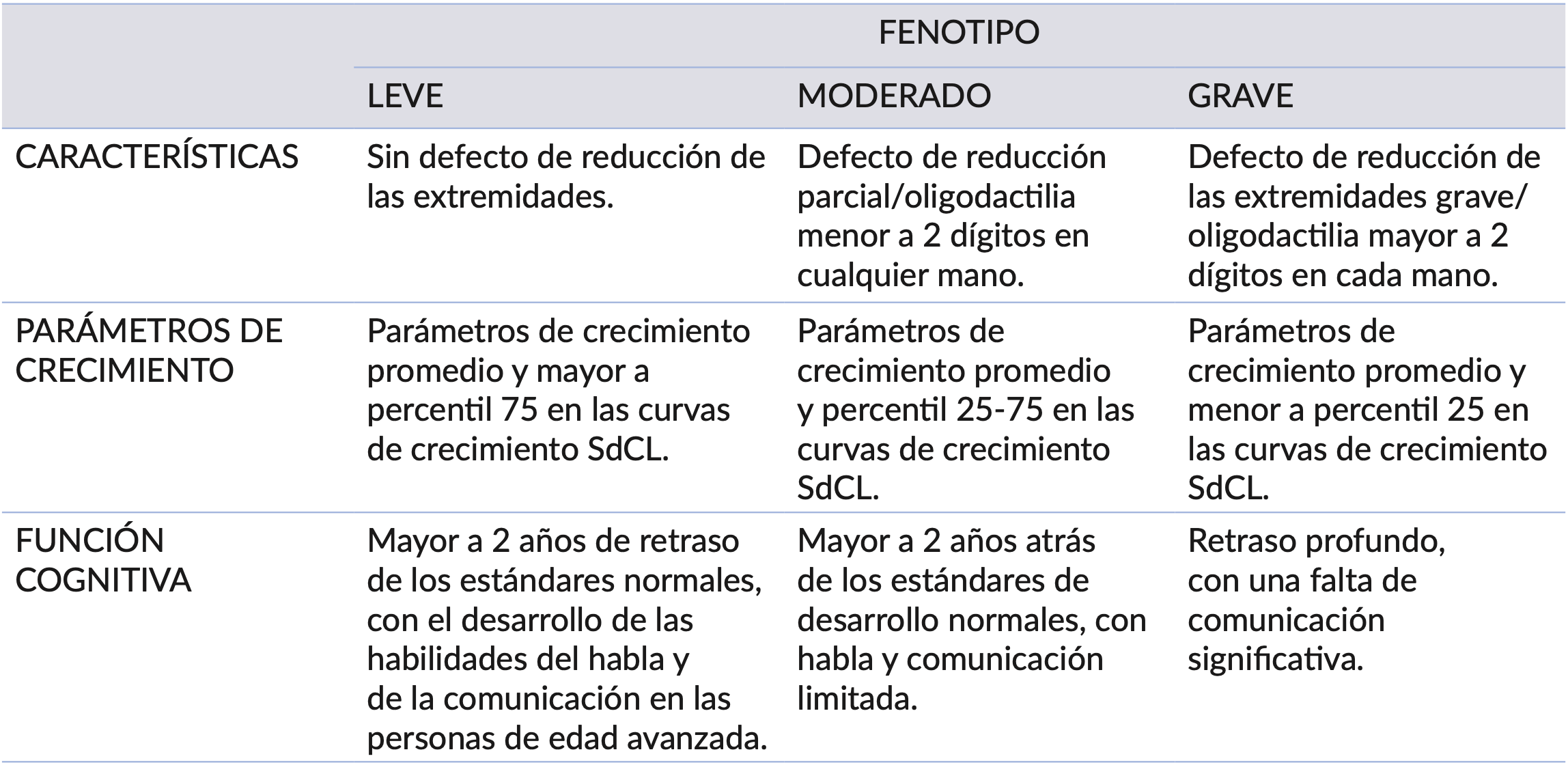
Al examen clínico general se observa cráneo braquimicrocefálico, retraso psicomotor, del lenguaje y características fenotípicas propias del síndrome, como hirsutismo generalizado, sinofridia con cejas arqueadas, pestañas largas y finas, nariz pequeña con puente nasal deprimido y ancho, narinas antevertidas y un filtrum alargado y prominente, labio superior fino con comisuras orientadas hacia abajo, implantación baja del cuero cabelludo, antihelix prominente, telecantus, pliegues epicánticos internos, pliegue palmar simio izquierdo, (Figura 1a). Se registra un peso de8kgytallade73cmalmomentode la consulta odontopediátrica. Según las tablas de percentil de la OMS para niñas se clasificó Edad-talla: Menor a 3, Peso-Edad: Menor a 3, Peso-Talla: 15.
Figura 1. a) Paciente con Síndrome de Cornelia de Lange en la primera consulta. b) Fotos tomadas en la primera consulta en donde se pueden observar rebordes gingivales superior e inferior con ausencia de unidades dentarias. c) Maxilar y Mandíbula con unidades dentarias que fueron erupcionando con retardo en las consultas de control.
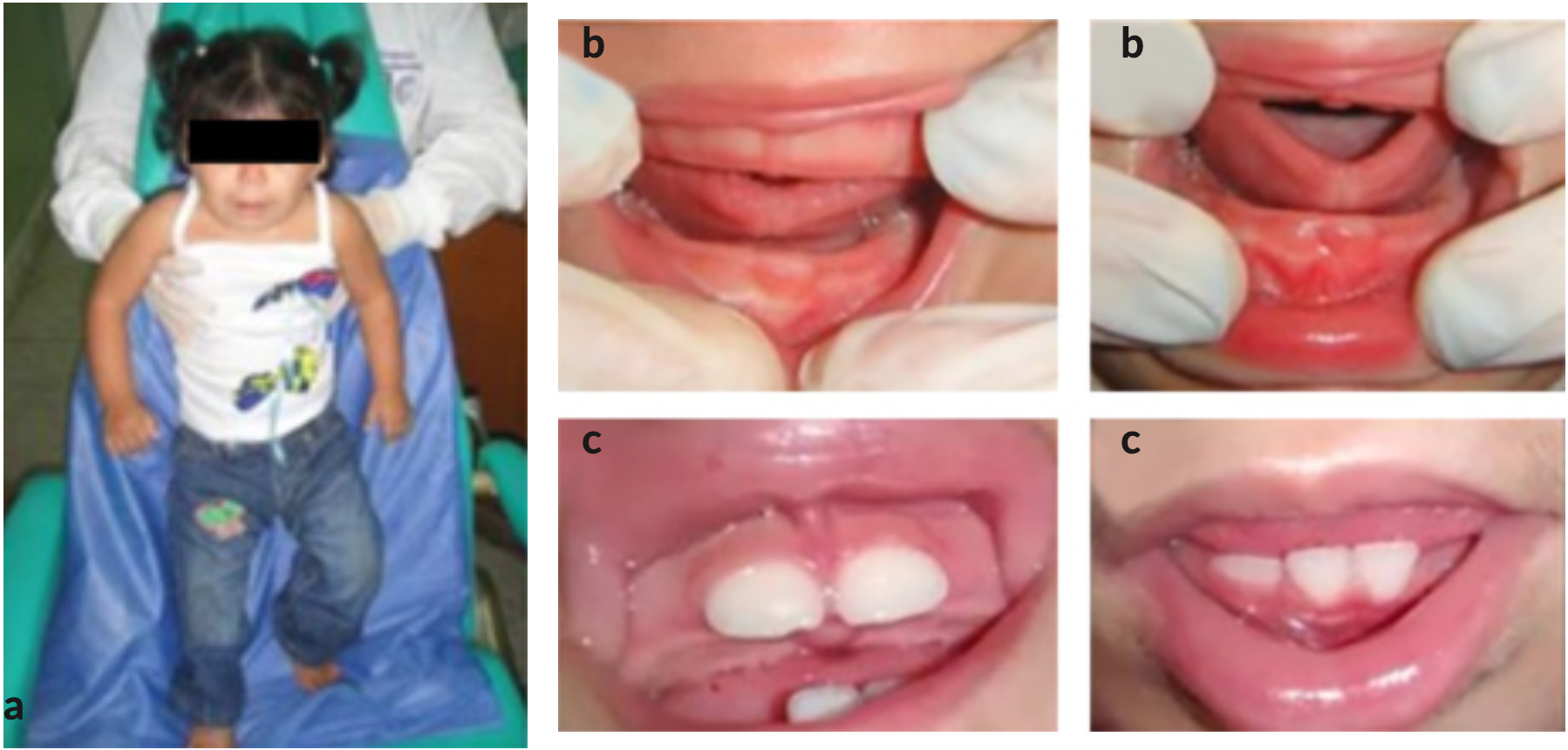
A la evaluación clínica intrabucal se observa: rebordes gingivales de color rosado, textura normal, inserción normal de frenillos, y ausencia de unidades dentarias, presentando un retardo de erupción de acuerdo a su edad y género, (Figura 1b). Como tratamiento se realiza registro fotográfico, prevención, educación para la salud bucal, medidas de higiene, orientación dietética y se indica realizar radiografías como examen complementario.
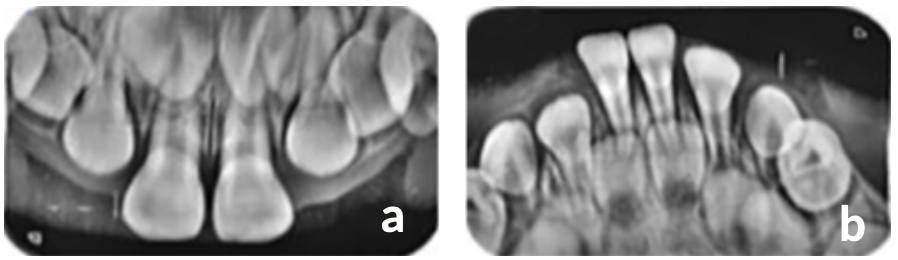
Una vez transcurrido 4 meses desde la primera consulta, se realizó una consulta control y se observó la erupción de incisivos centrales superiores e inferiores, (Figura 1c). Al realizar el examen radiográfico, se encontraron presencia de unidades dentarias 51, 52, 53, 61, 62 y 63 y se observó germen dental de unidades 11 y 21 en radiografía oclusal superior, (Figura 2a). Y en la radiografía oclusal inferior, presencia de unidades dentarias 81, 82, 83, 71, 72, 73 y 84, (Figura 2b).
Dado los hallazgos clínicos y radiográficos encontrados se observó claramente una alteración en la cronología y secuencia de erupción dentaria, puesto que erupciona el primer molar primario antes de terminar la erupción de todo el grupo incisivo, alterando así la secuencia de erupción. Así mismo la cronología se encontró con alteración, ya que la edad de la paciente no coincide con los estándares normales de cronología de erupción, que comienza a partir de los 6 meses de edad. El pronóstico de este caso es favorable, ya que se observó en el examen radiográfico la formación de los gérmenes dentales permanentes y las unidades dentarias primarias próximas a erupcionar, sin embargo se ve afectado el crecimiento de los maxilares, que en el futuro tendrá como consecuencia algún tipo de maloclusión.
Figura 2. Exámenes radiográficos (a. Rx oclusal superior, b. Rx oclusal inferior)
La erupción dentaria humana es un proceso de desarrollo único en el organismo, y los tiempos de erupción junto con la madurez ósea de un individuo presentan una correlación que siempre se debe considerar. Estudios han demostrado que los tiempos de erupción son específicos de género afirmando que los dientes de las niñas entran en erupción antes que los niños. Así mismo, existe una fuerte correlación entre el tiempo de erupción y la madurez dental. Los dientes normalmente erupcionan cuando han alcanzado 2/3 de la longitud de la raíz, sin embargo, existen síndromes en los cuales los dientes a pesar de haber alcanzado su longitud no erupcionan 27,28.
La edad ósea constituye el indicador de madurez biológica más útil para caracterizar ritmos o “tiempos” de maduración durante el crecimiento, ya que otros indicadores tradicionalmente utilizados, se limitan a ciertas etapas de la vida y muestran gran variabilidad, en especial durante la pubertad29.
Por esta razón, la verdadera edad biológica de un individuo durante su crecimiento sólo se puede obtener de su edad ósea, y estimar desde el período neonatal hasta el final del crecimiento. Según Espinosa (2011), la edad cronológica es la que va desde el nacimiento hasta la edad actual de la persona. La edad biológica, tiene en cuenta los cambios físicos y biológicos que se van produciendo en las estructuras celulares, de tejidos, órganos y sistemas29.
Entonces la edad dental es considerada un indicador confiable de la edad cronológica y ha sido utilizada tanto en la práctica odontológica, con la finalidad de determinar si la maduración dental del paciente está dentro del promedio para su grupo de edad30.
En relación con los estudios que se han venido realizando, un caso revela que se presenta retraso en la edad esquelética (4 y 6 meses) y confirma un 40% de retraso en el crecimiento y retardo de la erupción en un paciente pediátrico, y se relaciona con otros estudios coincidiendo en que se presenta retardo de la erupción en dentición primaria y en el crecimiento óseo6.
Cabe destacar que Mehta, en un reporte de caso de un paciente masculino de 10 años de edad con diagnóstico de SdCL señala retardo de erupción, maloclusión, apiñamiento, micrognatia, filtrum largo, labio superior delgado y caries dental, destacando una vez más la alteración de cronología de erupción en dicho síndrome14.
En este orden de ideas se puede citar otro caso de un paciente masculino de 12 años de edad con diagnóstico genético de SdCL que presenta retardo de erupción tanto de la dentición primaria como permanente, por lo tanto esta característica coincide con estudios ya descritos y se correlaciona con el diagnóstico del caso que estamos reportando 20.
De la misma forma diversos autores coinciden con reportes de retardo de erupción en casos de síndrome de Cornelia de Lange, algunas anormalidades dentales reportadas incluyen erupción retardada, diastemas, macro y microdoncia (17). Yamamoto y et al, en su investigación, han informado dos casos con erupción dental tardía y microdoncia 31.
También se realizó un estudio reciente en donde se revisaron de forma sistemática 7 casos publicados de SdCL y la manifestación bucal que más se repite es la erupción dentaria tardía y por consiguiente; se presentan maloclusiones, malposición dentaria, caries dental y enfermedad periodontal 32.
En resumen, se encuentra que posibles agentes causales de la erupción dentaria, incluyen: influencia endocrina, cambios vasculares, y degradación enzimática31. Por lo tanto se puede decir que síndromes y enfermedades sistémicas en las cuales estos agentes son afectados, podrían generar retardo de erupción dentaria. Finalmente, es pertinente acotar que todo lo anteriormente descrito tiene similitud con el caso clínico estudiado y tratado en el postgrado.
De acuerdo a los últimos reportes la cronología de la erupción también se puede ver afectada por factores sistémicos, patologías endocrinas, radiación o síndromes, tanto así que cada día se reportan nuevos síndromes craneofaciales y en muchos de ellos se observa como características importantes los desórdenes en la erupción dental.
Como se ha mostrado en la literatura, el retardo de la erupción es uno de los signos patognomónicos que se muestran en el Síndrome de Cornelia de Lange, por lo tanto se concluye que el presente caso clínico coincide con estudios ya descritos donde refieren que el retardo de la erupción se ha presentado en pacientes de diferentes edades con diagnóstico genético de SdCL, presentando tanto alteración de la cronología como secuencia de erupción.
Este retardo de erupción puede tener correlación con la madurez ósea, ya que dicho síndrome presenta retardo del crecimiento desde la formación intrauterina hasta después del nacimiento. Sin embargo todos los estudios sobre la erupción dental concluyen que el proceso de erupción o el mecanismo detrás de la erupción dentaria debe ser investigado a profundidad, por lo que se recomienda ampliar los estudios sobre la erupción dentaria y su relación con los síndromes que afectan el crecimiento craneofacial incluyendo el síndrome de Cornelia de Lange.