 , Luis Eduardo Carmona Arango2, Ángel Castro Dager3
, Luis Eduardo Carmona Arango2, Ángel Castro Dager3DOI:https://doi.org/10.47990/alop.v11i2.324
Sandra Viviana Caceres Matta1, Luis Eduardo Carmona Arango2, Ángel Castro Dager3
1Sandra Viviana Caceres Matta, Odontologa. MSc Bioquimica Clinica,
Residente de primer año Especialidad de Odontopediatria y Ortopedia Maxilar, Docente Escuela de Odontología
Universidad del Sinú – Seccional Cartagena, Grupo de Investigacion PROMOUC.
2Luis Eduardo
Carmona Arango, Odontologo, Especialista en Odontopediatria, MSc. Microbiología, Docente Facultad de
Odontologia, Universidad de Cartagena, Director Grupos de Investigación PROMOUC.
3Ángel
Castro Dager, Docente Facultad de Medicina, Universidad de Cartagena Unidad Hemato-oncología Pediátrica.
Universidad de Cartagena. Clínica Blas de Lezo, Cartagena, Colombia.
La Mucolipidosis tipo II es una enfermedad autosómica trastorno recesivo clínicamente caracterizado por la dismorfia facial y una hiperplasia gingival severa. Relato del caso: Se reporta caso de paciente de 2 años de edad, con diagnóstico de enfermedad metabólica tipo mucolipidosis II. Al examen físico se encontraron facies tosca, marcada hiperplasia gingival sintomática generalizada en maxilar superior e inferior, encías sangrantes, cuello corto, con regular sostén cefálico, piel delgada, pectus excavatus, codos normales, manos con disminución en el agarre y piel gruesa con xerosis, dificultad para elevar los brazos por encima de la cabeza, retardo global en neurodesarrollo. Siendo así, se manejó el caso de manera multidisciplinaria, permitiendo que el paciente evolucione de manera positiva al tratamiento integral, con mejoramiento en la motricidad. Conclusiones: Los fenotipos clínicos superpuestos son un desafío de diagnóstico para el personal de la salud en Odontología, especialmente en casos de mucolipidosis (ML) y trastornos mucopolisacáridos (MPS), debido a la superposición de las características clínicas.
Palabras clave: Mucolipidosis, diagnóstico, mutación, hiperplasia gingival, lisosomal.
A mucolipidose tipo II é um distúrbio autossômico recessivo caracterizado clinicamente por dismorfia facial e hiperplasia gengival grave. Relato de caso: É relatado o caso de uma paciente de 2 anos com diagnóstico de doença metabólica do tipo mucolipidose II. O exame físico revelou fácies grosseira, hiperplasia gengival sintomática generalizada acentuada na mandíbula superior e inferior, gengivas sangrantes, pescoço curto, com apoio de cabeça regular, pele fina, pectus excavatus, cotovelos normais, mãos com pega diminuída e pele grossa com xerose, dificuldade em levantar os braços acima da cabeça, atraso no desenvolvimento neurológico global. Portanto, o caso foi tratado de forma multidisciplinar, permitindo que o paciente evoluísse de forma positiva para um tratamento integral, com melhora nas habilidades motoras. Conclusões: A sobreposição de fenótipos clínicos é um desafio diagnóstico para o pessoal de saúde em Odontología, especialmente nos casos de mucolipidose (ML) e distúrbios dosmucopolissacarídeos(MPS),devidoà sobreposição de características clínicas.
Palavra-chave: Mucolipidose, diagnóstico, mutação,hiperplasiagengival,lisossomal.
Mucolipidosis type II is an autosomal recessive disorder clinically characterized by facial dysmorphia and severe gingival hyperplasia. Case report: The case of a 2-year-old patient with a diagnosis of metabolic disease type mucolipidosis II is reported. Physical examination revealed coarse facies, marked generalized symptomatic gingival hyperplasia in the upper and lower jaw, bleeding gums, short neck, with regular head support, thin skin, pectus excavatus, normal elbows, hands with decreased grip, and thick skin with xerosis, difficulty raising the arms above the head, global neurodevelopmental delay. Therefore, the case was handled in a multidisciplinary way, allowing the patient to evolve in a positive way to comprehensive treatment, with improvement in motor skills. Conclusions: Overlapping clinical phenotypes are a diagnostic challenge for health personnel in Dentistry, especially in cases of mucolipidosis (ML) and mucopolysaccharide disorders (MPS), due to the overlapping of clinical characteristics.
Key words: Mucolipidosis, diagnosis, mutation, gingival hyperplasia, lysosomal. (MeSH).
La mucolipidosis tipo II alfa / beta (ML II alfa / beta; OMIM # 252500) es una enfermedad rara autosómica recesiva dentro de los errores innatos del metabolismo causado por la actividad reducida de la N-acetilglucosamina-1-fosfotransferasa (GlcNAc-1-fosfotransferasa; EC 2.7.8.17) debido a las mutaciones en el gen GNPTAB (NG021243; GeneID 79158). GlcNAc-1-fosfotransfereasa cataliza el primer paso en la generación de manosa 6-fosfato, se requiere un marcador de reconocimiento para un direccionamiento eficiente de enzimas lisosomales solubles en los lisosomas. Defectos en GlcNAc- 1-fosfotransferasa conducen a la mala distribución de varias enzimas lisosómicas y a la acumulación de macromoléculas en a nivel lisosomal no degradadas.1 Dentro los hallazgos patológicos microscópicos reportados en estas células defectuosas se encuentran inclusiones citoplasmáticas formadas por la acumulación de varias macromoléculas, lo cual desarrolla a nivel clínico un fenotipo típico caracterizado por rasgos faciales gruesos, hiperplasia gingival, disostosis múltiple, contracturas articulares y muerte a una edad temprana.2 Actualmente no existe un tratamiento específico para ML II alfa / beta, aunque el trasplante de células hematopoyéticas se puede intentar en casos diagnosticados muy temprano.3 Al ser un trastorno autosómico recesivo, los individuos heterocigotos para ML II alfa / beta no se ven clínicamente afectados, pero pueden exhiben anormalidades bioquímicas intermedia.
La mucolipidosis tipo II, es una patología debilitante progresiva, con crecimiento osteomuscular reducido y obstrucción en el desarrollo posterior a cumplir los primeros dos años de vida.3 De esta manera, el desarrollo psicomotor es lento, gran porcentaje de estos pacientes no logran caminar de manera independiente. Entre las características se encuentra hernia umbilical, anomalías a nivel cardiaco específicamente en las válvulas, rasgos faciales “toscos”, hiperplasia gingival, endurecimiento de las cuerdas vocales, vía respiratoria estrecha, estrabismo manifiesto e hipertelorismo, manos en forma de garra, encurvamiento de la columna vertebral (Figura 1) y de los miembros inferiores, este tipo de afecciones multisistemicas no sobreviven más allá de la primera década de infancia.4,5
La sospecha clínica es el primer paso en el diagnóstico de ML II alfa / beta. El diagnóstico definitivo depende en la demostración de baja actividad de GlcNAc- 1-fosfotransferasa utilizando el [32P] UDP- GlcNAc radiactivo sustrato. Sin embargo, como este ensayo solo está disponible en muy pocos laboratorios, por lo general, se establece un diagnóstico indirecto mediante la medición de hidrolasas lisosomales células mesenquimales donde sus niveles deben ser bajos y a nivel del entorno extracelular circundante sus niveles deben ser altos.6 Aunque generalmente no se requieren pasos adicionales y el análisis genético GNPTAB generalmente no es necesario para confirmar el diagnóstico, las pruebas de ADN son útiles cuando las pruebas bioquímicas no son concluyentes o en el caso de diagnosticar portadores de la afección. El diagnóstico prenatal de ML II alfa / beta se puede establecer mediante el mismo enfoque bioquímico, evaluando material fetal obtenido de biopsia de vellosidades coriónicas, amniocentesis o cordocentesis.7 Asimismo, el ADN puede extraerse y aislarse de estos materiales fetales y analizarse como en el período postnatal, aunque este enfoque basado en el ADN prenatal requiere información adicional sobre el genotipo parental o el flujo / naturaleza de alelos causantes de la enfermedades.8
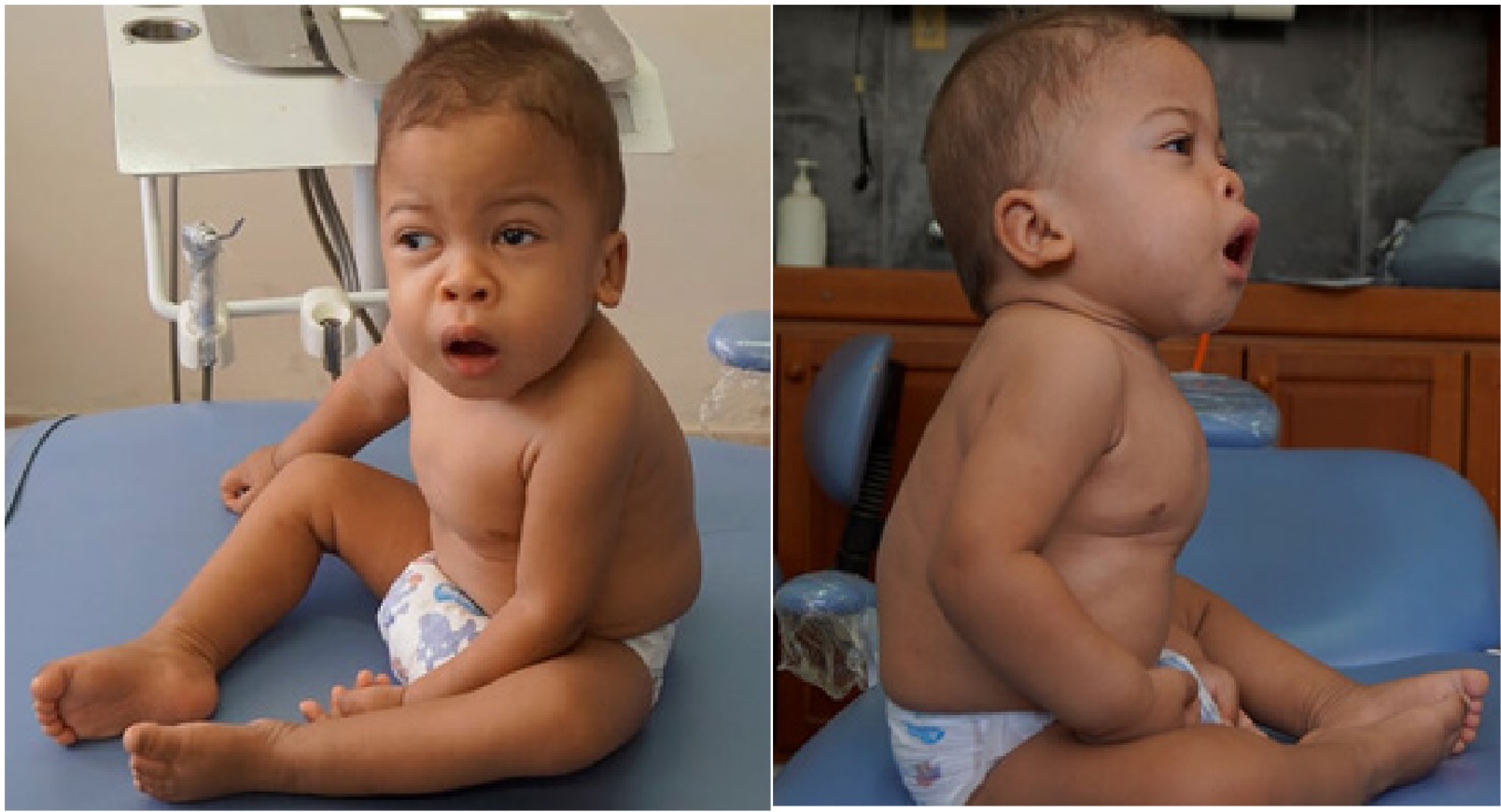
Figura 1: Paciente afectado con Mucolipidosis tipo II o células de inclusión, donde se evidencia las facies toscas características de la condición. Paciente respirador oral por la hiperplasia gingival severa.
La prevalencia de la mucolipidosis tipo II es aproximadamente 1 de 500.000 nacidos vivos a nivel mundial. Específicamente se ha estimado en 1 / 123.500 nacidos vivos en Portugal, 1 / 252.500 en Japón y 1 / 625.500 en los Países Bajos.9,10 En Colombia se identifican alrededor de 1920 enfermedades huérfanas según datos reportados por las diferentes entidades de salud en el país, donde se incluye la mucopolidosis tipo II como una de estas, los según la resolución 430 del 2013.11
Se realiza socialización de consentimiento y asentamiento informado a la madre del menor donde también se anexa párrafo para el consentimiento fotográfico, se procede a realizar historia clínica con sus anexos. Paciente de género masculino de 26 meses de edad, quien al nacer fue hospitalizado por complicaciones respiratorias en unidad de cuidados intensivos neonatal durante 11 días. Durante el primer semestre de vida el niño evolucionó con hipotonía moderada y leve retraso del desarrollo psicomotor y pondoestatural. Se hospitalizó en varias ocasiones por presentar trastornos respiratorios. Desde los 7 meses se observó además peso y talla estacionarios, progresión de las dismorfias faciales y esqueléticas y deterioro sicomotor. A los 12 meses el menor presenta episodios febriles intermitentes, sin una causa infecciosa documentable. Motivo por el cual fue remitido a pediatría y a odontopediatria al presentar bajo peso y retardo en el desarrollo sicomotriz de acuerdo a su edad al igual que hiperplasia gingival, se realizó manejo interdisciplinario, al igual que se ordenan exámenes paraclínicos (Tabla 1 y 2). Posterior a la valoración por las dos especialidades, se solicita interconsulta con la especialidad de genética, por sospecha clínica de mucopolisacaridosis tipo I, de acuerdo a la solicitud se analizaron los siguientes genes correspondientes al panel NGS mucopolisacaridosis (4 genes) GNPTAB, GNPTG, MCOLN1 y NEU1. En los resultados se obtiene: GNPTAB: c.3503_3504delTC(p.Leu1168Glnfs*5), Frameshift patogénica. En los datos de secuencia del panel génico se detectaron 14 variantes de secuencia y dentro de ellas se identificó una variante de tipo frameshift en el gen GNPTAB.
Tabla 1: Actividad enzimática.
| Arilsulfatasa A en leucocitos | Valor de paciente 13.9mmol/h/mg control: 36,3 mmol/h/mg |
| Arilsulfatasa A en suero | Valor de paciente 376 mmol/h/mg control: 43,9 mmol/h/mg |
| Glucosaminoglucanos | Creatinuria mix aislada: 10 mg/dl , Creatinuria mix aislada 2: 10 mg/dl, Creatinuria mix aislada 3: 23 mg/dl, Creatinuria mix aislada 4: 54 mg/dl (errores innatos) |
| Alfa L iduronidasa | 51,46 mmol/ml/hr (NORMAL) |
| Aril sulfatasa B | 85,04 mmol/ml/hr (NORMAL) |
| IDS | Sin variante patogenica Normal |
| Esfingomielinasa Acida | > 71,2 uMol/h (límite superior) |
| Iduronato sulfatasa | > 74,0 uMol/h (limite superior) |
| Arilsulfatasa A en leucocitos | Valor de paciente 13,9 mmol/h/mg control: 36,3 mmol/h/mg |
| Arilsulfatasa A en suero | Valor de paciente 376 mmol/h/mg control: 43,9 mmol/h/mg |
| Glucosaminoglucanos | Creatinuria mix aislada: 10 mg/dl , Creatinuria mix aislada 2: 10 mg/dl, Creatinuria mix aislada 3: 23 mg/dl, Creatinuria mix aislada 4: 54 mg/dl (errores innatos) |
| Alfa L iduronidasa | 51,46 mmol/ml/hr (NORMAL) |
| Aril sulfatasa B | 85,04 mmol/ml/hr (NORMAL) |
| IDS | Sin variante patogenica Normal |
| Esfingomielinasa Acida | > 71,2 uMol/h (límite superior) |
| Iduronato sulfatasa | > 74,0 uMol/h (limite superior) |
Tabla 2: Reporte de exámenes complementarios.
| Estudio | Resultado |
| Hb | 11,3 gr/dl |
| Hto | 35% |
| Leucocitos | 13.300 |
| PMN | 30,7% |
| Linfocitos | 57,9% |
| Monocitos | 8,0% |
| Eosinófilos | 3,2% |
| Basófilos | 0,03% |
| Cariotipo | 46 XY |
| Panel NGS | Mutación gen GNPTAB |
| Estudio | Resultado |
| Rx columna lumbosacra | Platispondilia, defecto en fusión arcos posteriores l4, l5 y s1, aumento en sifosis dorsal, engrosamiento de la cavidad medular en arcos costales, coxa magna en pelvis, leve osteopenia |
| Tac de tórax simple | Defecto diafragmático retroesternal con dmt ap de 2,5 cm corazón normal, tráquea, bronquios parénquima pulmonar normal, osteopenia difusa |
| Rx columna cervical | Pobre definición de espaciones vertebrales, acortamiento del cuello, disminución de espacios intervertebrales |
| Rx digital de huesos largos serie completa | Cráneo: Ligera asimetría craneofacial con suturas y fontanelas poco acentuadas para la edad, silla turca normal. Huesos largos: ligera desmineralización ósea, relaciones articulares conservadas, ejes centrados, ensanchamiento en huesos largos, deformidad discreta de los mismos, cavidades coxales deformadas con alerones ilíacos ensanchados |
| Ecografía de abdomen total | Hígado con volumen, morfología y ecogenicidad normales sin lesiones difusas, vesícula y biliar normal, páncreas y bazo ecogenicidad normales, riñones: forma y tamaño conservado homogéneo sin lesiones quísticas, no se observa líquido libre, vejiga urinaria volumen y forma normal. |
| Rx senos paranasales | Rx panorámica: poca neumatización de senos maxilares que coincide con edad. |
| Tac de cara | Pendiente realización |
| Rx cara lateral de cráneo fotografía 2 | No se observa alteración del seno esfenoidal |
| Potenciales evocados auditivos y visuales | Auditivo: se obtienen ondas I-III y V con baja amplitud bilateral Visuales: se obtiene respuesta N1-P100-N2 con morfología, amplitud y latencias limítrofes para la edad. |
| Tp | 14,8 seg |
| Tpt | 34,2 seg |
Se identificó en el paciente la mutación c.3503_3504delTC (NM_024312) en estad homocigoto, la cual se encuentra reportada en las bases de datos Ensemble, NCBI y ExAc (rs34002892). Su frecuencia alélica es menor del 1% (MAF<0,01), lo cual sugiere que el cambio descrito puede tener un impacto negativo en le fenotipo, dada su baja frecuencia en la población en general. La mutación frameshift c.3503_3504delTC consiste en la deleción de las bases timina y citosina, lo que causa el corrimiento en el marco de lectura, generando una proteína truncada y probablemente no funcional, con una cola aberrante de 4 residuos y un codón de parada prematuro (p.Leu1168Glnfs*5). Según la base de datos CliVar, su significancia es patogénica.
Dada la profundidad de la variante identificada respecto a la profundidad total (138X/192XX), se realizó confirmación de la misma mediante secuencia de Sanger. En la figura 2 se puede apreciar la variante c.3503_3504delTC en el estado homocigoto.
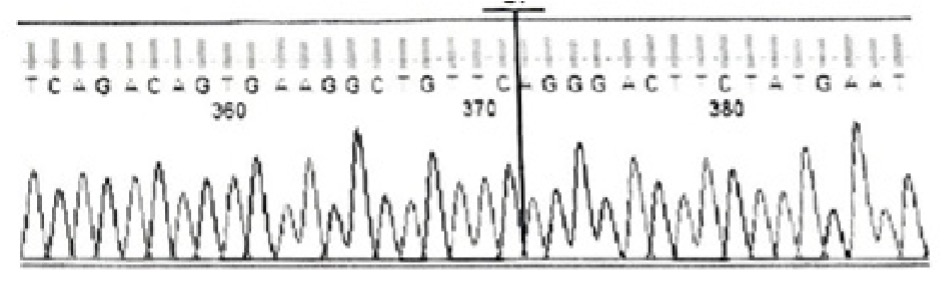
Figura 2: Electroferograma que indica la presencia de la variante homocigota c.3503_3504delTC del gen GNPTAB.
Dado el potencial impacto de las mutaciones tipo frameshift, que pueden llevar a la perdida de la función de la proteína y con base en la evidencia científica, la mutación homocigota GNPTAB: c.3503_3504delTC (p.Leu1168Glnfs*5) está clasificada como patógena para mucolipidosis tipo II alfa/beta. Dentro de los exámenes complementario se realizó cariotipo al paciente, donde se reporta 46XY, no se identificaron alteraciones cromosómicas estructurales o numéricas (Figura 3). Al examen físico presentó peso 9,5 Kg, estatura 76 cms con disminución en la curva del desarrollo de - 3 (Según curvas de patrón de crecimiento de la OMS 2009) (Figura 4), piel seca generalizada, textura y facies gruesas, disminución de la hendidura palpebral, estrabismo horizontal, hipertelorismo, reflejo pupilar aferente disminuido, cuello corto, fondo de ojo normal. Pobre sostén cefálico sin adenopatías, conductos auditivos regulares, hiperplasia gingival, apertura de la boca en posición de reposo, con respiración oral (Figura 5). Tórax excavatum expansible sin retracciones costales, cifosis dorsolumbar, deformidad toraxica, hernia umbilical con anillo de aproximadamente 2 cms coercible, no encarcelada, limitación para la movilización articular en los miembros superiores e inferiores, hombros no sobrepasan la cabeza, codos que logran extenderse completamente, rodillas y dedos de las manos de predominio derecho, mano izquierda con los dedos en flexión (mano en garra) (Figuras 6, 7), resto del examen sistémico fue normal.
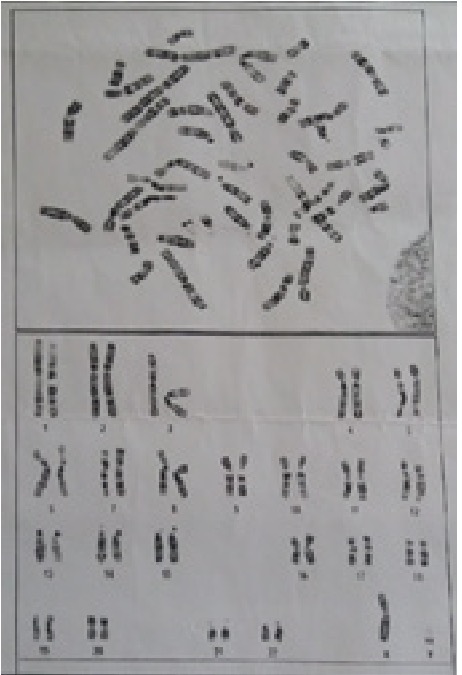
Figura 3: Cariotipo al paciente, donde se reporta 46XY, no se identificaron alteraciones cromosómicas estructurales o numéricas.
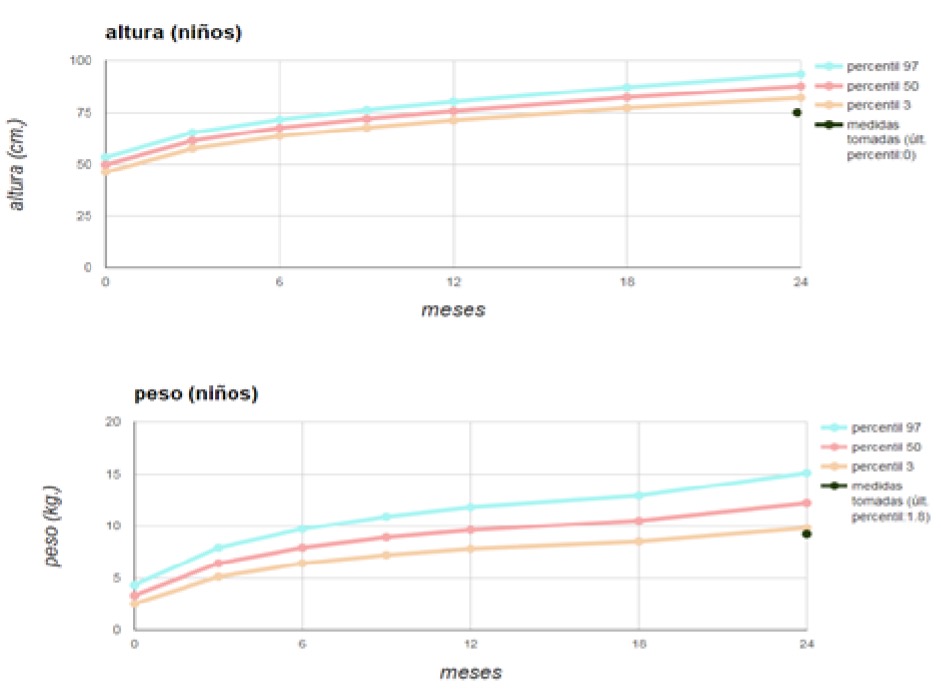
Figura 4: Patrones de crecimiento altura y peso.
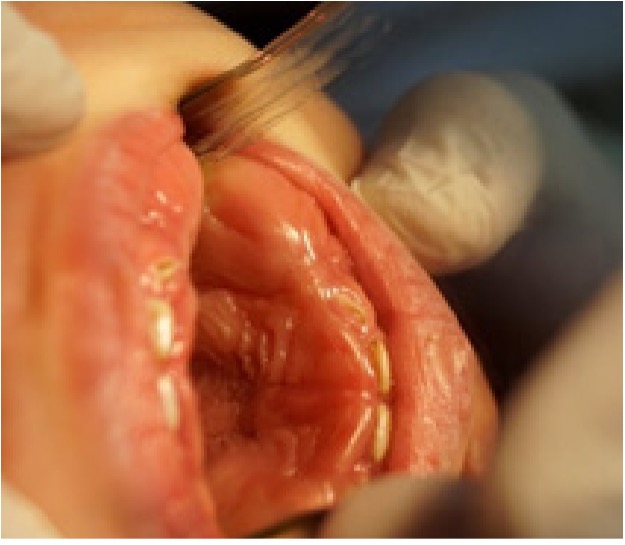
Figura 5: Fotografía de Hiperplasia Gingival, se evidencia leve erupción del tercio incisal de los órganos dentarios antero-superiores.
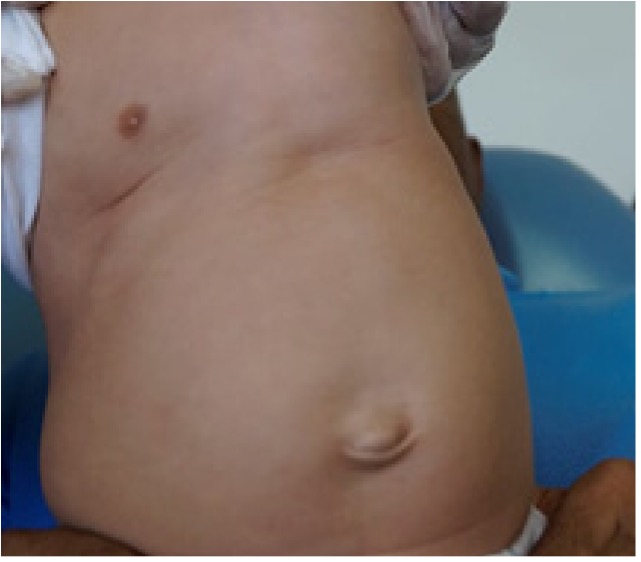
Figura 6: Fotografía de hernia umbilical
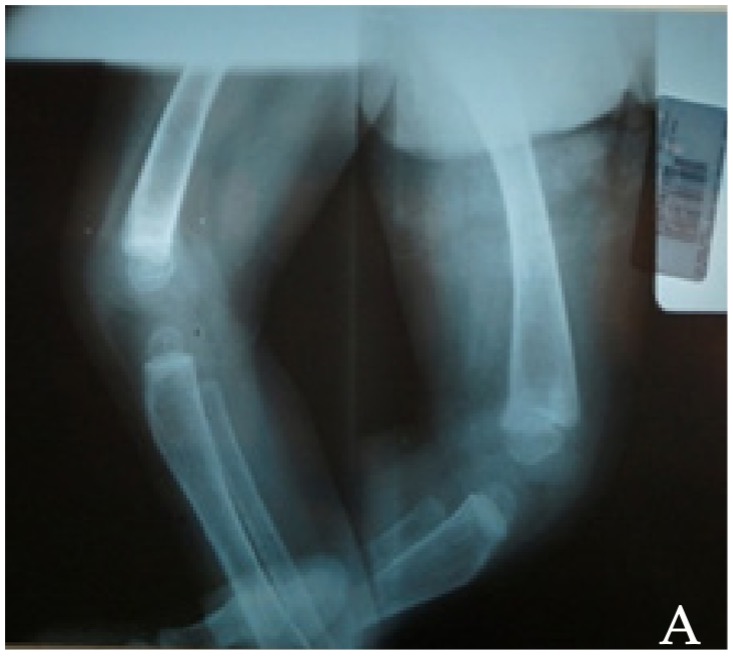

Figura 7: A. Curvamiento de los miembros inferiores. B. Radiografía de Columna y Tórax. Radiografía de columna lateral de la paciente con mucolipidosis II. Esta radiografía demuestra una xifosis dorsal alta angulada, cuerpos vertebrales biconvexos y con acortamiento antero-posterior y aguzamiento de rebordes antero-inferiores de cuerpos vertebrales lumbares.
Al examen intraoral se evidenciaron manchas extrínsecas de color verde- amarillentas a nivel de órganos dentarios 52, 51 y 61 en tercio medio de la cara vestibular, causados por la ingesta de sulfato ferroso desde los seis meses de edad, al igual que lesión cariosa tipo 2B según código ICDAS a nivel de cara vestibular de órgano dentario 61, de textura rugosa a la palpación con la sonda periodontal, se calcula un índice de placa bacteriana elevado, el cual fue tratado con cambio de hábitos orales, educación y cuidados indicados a la madre. Paciente presenta a nivel de labio superior e inferior queilitis generalizada. Paciente presenta hábito de succión digital, lo cual agrava la condición de vestibularización de los órganos dentarios incisivos superiores. Igualmente, el paciente presenta incompetencia labial, respirador oral (Figuras 8. A, B). De acuerdo a los hallazgos radiográficos y clínicos paciente presenta asimetría facial en la primera consulta debido a lesión tumoral lobulada de aproximadamente 5 cm de diámetro, 2 cm de alto, de color violáceo brillante, de consistencia pétrea a la palpación, sangrante, sintomática de aproximadamente dos semanas de evolución, la cual le imposibilitaba la alimentación y cierre bucal debido a su tamaño al paciente (Figuras 9, 10) . Posterior a la valoración radiográfica y clínica por Odontopediatría se decide tratar estar lesiones con aspirados de líquido capsular de la lesión, con seguimiento clínico cada 15 días hasta la fecha.
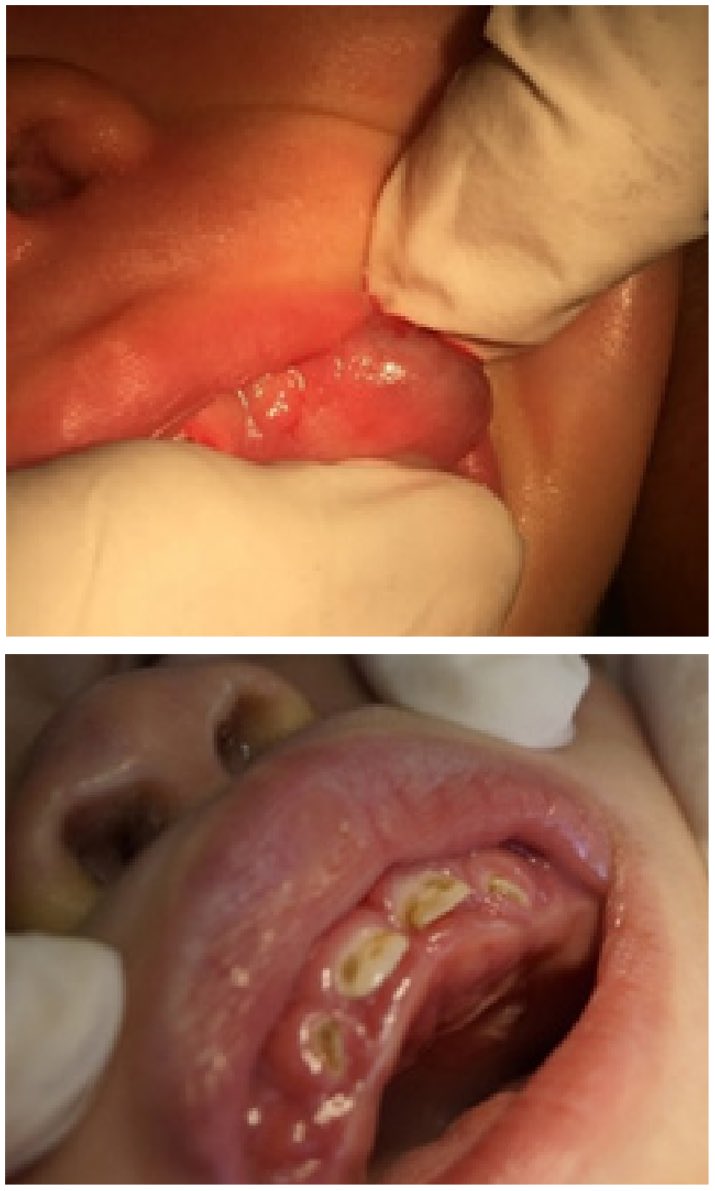
Figura 8: A. Se evidencia lesión tumoral a nivel de maxilar superior izquierdo de aproximadamente 5 cm de diámetro, 2 cm de alto, de color violáceo brillante, de consistencia pétrea a la palpación, sangrante, sintomática de aproximadamente dos semanas de evolución, la cual le imposibilitaba la alimentación y cierre bucal debido a su tamaño al paciente. B. Paciente presenta hiperplasia gingival generalizada, la cual impide la erupción normal de las piezas dentarias, paciente respirador oral por la hipertrofia de las estructuras orales, se evidencia paladar estrecho con poco desarrollo a nivel clínico y radiográfico, rugas palatinas normales. Manchas hipercrómicas a nivel de los órganos dentarios 51, 52, 61 y 62 a nivel de tercio medio de cara vestibular.
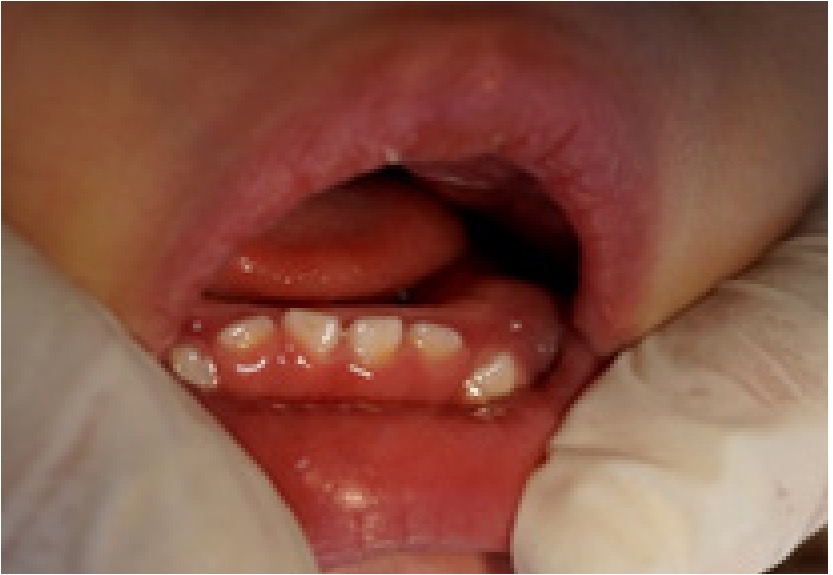
Figura 9: Fotografía maxilar inferior con hiperplasias gingivales, mal posición de órganos dentarios antero inferior.
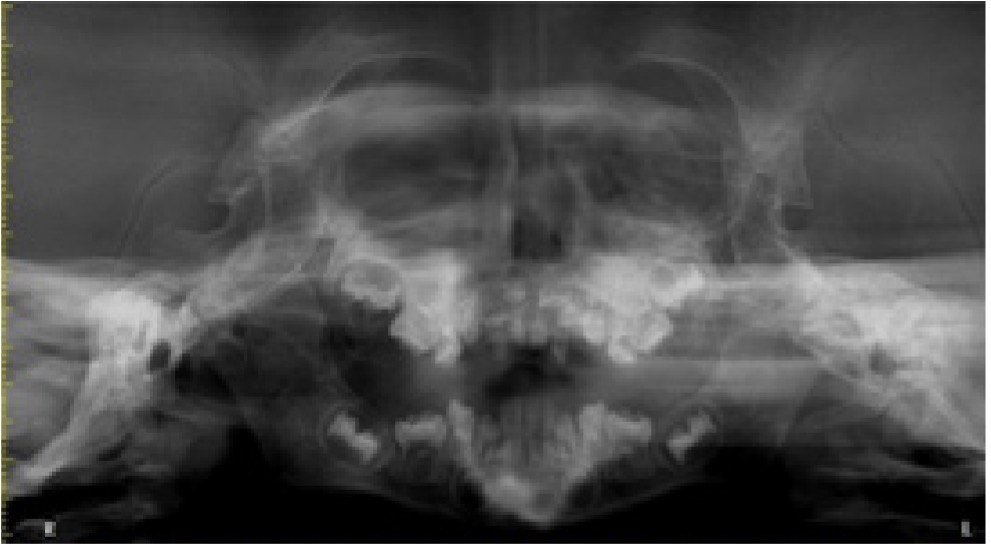
Figura 10: Radiografía panorámica, se observa poco desarrollo a nivel transversal de los maxilares superior e inferior del paciente, se observa una hipertrofia de los senos paranasales.
Radiográficamente se evidencia paladar estrecho con poco desarrollo a nivel clínico. Los exámenes complementarios con las radiografías panorámicas y TAC de Cabeza y cuello, evidenciaron: una cortical mandibular engrosada, folículos densamente agrandados asociados con las coronas de los primeros molares permanentes, los cuales se encontraban deprimidos cerca del borde inferior de la mandíbula, se evidenció poco desarrollo bilateral de los cóndilos mandibulares (Figuras 11,12).
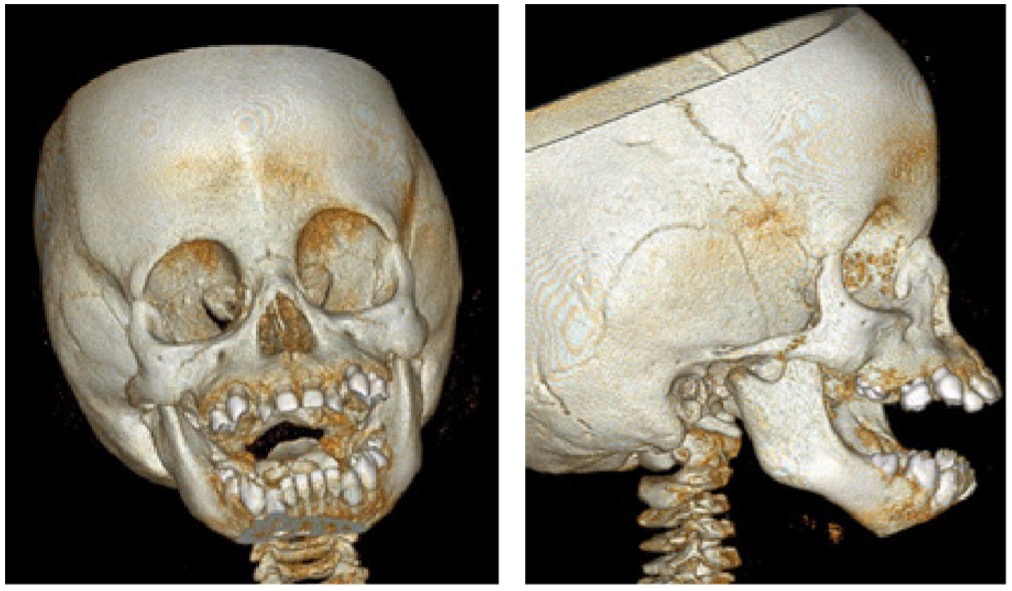
Figuras 11 y 12: Radiografía 3D lateral de cráneo. Fotografía lateral de hemifacie derecha de características semiológicas se evidencia paciente respirador oral, por el poco desarrollo de los maxilares y la mal posición dentaria.
Se reporta caso de niño en el que se confirmó el diagnóstico de una Mucolipidosis tipo II. Las mucopolisacáridos y mucolipidosis presentan un patrón fenotípico similar de rasgos faciales grotescas, hiperplasia gingival, retraso sicomotriz, encurvamiento de los miembros superiores e inferiores y cuello corto; el estudio de la actividad enzimática y genética es el hallazgo confirmatorio del diagnóstico.12,13 Entre las características más relevantes de la mucolipidosis II, está el fenotipo hurleriano, definido con viceromegalia y disostosis múltiple. Si la vida del paciente se prolonga, se muestran mioclonías y mancha rojo cereza.
En las mucopolisacáridos el cuadro clínico es evidente por lo general a partir del segundo o tercer año de vida.
Otras enfermedades lisosomales que se pueden manifestar en forma similar durante el primer semestre de vida por un fenotipo hurleriano son la sialidosis y la galactosialidosis. El análisis de oligosacáridos en la orina y el estudio enzimático permiten en estos casos establecer la etiología correcta. Las características clínicas del niño presentado corresponden plenamente a las descritas para esta patología, coincidiendo la edad de inicio y la presencia de un deterioro psicomotriz rápidamente progresivo y de un fenotipo muy semejante al de la enfermedad de Hurler.14 En este caso, el diagnóstico preciso permitió entregar a los padres un consejo médico genético adecuado y oportuno. Se informó a los padres que ambos son portadores sanos para mucolipidosis tipo II y que la posibilidad de recurrencia en un nuevo hijo en común es 25%. Este tipo de afección es considerada como rara, por tener una prevalencia baja, debido a que los signos y síntomas no están siempre presentes desde el nacimiento, a excepción de los grados más severos de la misma, su diagnóstico se realiza a través del tiempo y a medida que las manifestaciones clínicas son evidentes y que las pruebas de laboratorio e imágenes apoyen la impresión diagnostica,15 Uno de los primeros indicios de esta condición es el dimorfismo facial y a nivel músculo esquelético, retardo en el desarrollo neurosicomotor, hipotonía generalizada, hiperplasia gingival lo cual perpetua la baja ingesta de alimentos pues limita la masticación y obliga al paciente ser respirador oral, provocando así la apertura de manera permanente, al igual que baja talla o peso con anomalías radiológicas severas.16 Desde el punto de vista radiológico, el diagnóstico diferencial incluye el síndrome de Hurler; la gangliosidosis GM1 tipo 1; la forma infantil de galactosialidosis; la sialidosis tipo 2; la enfermedad de depósito de ácido siálico libre, forma infantil; el raquitismo hipocalcémico y la Mucolipidosis tipo III. Distinguir radiológicamente entre una mucolipidosis tipo II y la enfermedad de Hurler puede ser difícil, ya que existen, varias alteraciones esqueléticas comunes a ambas entidades. Algunos hallazgos diferenciales entre ambas patologías son los fenómenos osteopáticos presentes en la Mucolipidosis tipo II, especialmente durante el primer año de vida. La osteopenia generalizada, con un patrón trabecular tosco y alteraciones metafisiarias típicas, presentes ya en el período neonatal, y que lo llevan a confundirse con el raquitismo.17 Sin embargo, en la ML II estas alteraciones tienden a regresar a fines del primer año de vida, dificultándose desde entonces su diferenciación con la enfermedad de Hurler.18 El diagnóstico diferencial con la gangliosidosis GM1 puede ser complejo durante el primer año de vida, más aún en el período de recién nacido, pues en esta etapa ambas patologías presentan fenómenos de periostitis.15 Sin embargo, en la gangliosidosis GM1, a diferencia de la ML II, estos fenómenos persisten a lo largo de la evolución de la enfermedad. La ML II comparte con el hiperparatiroidismo algunas de las alteraciones osteopáticas antes descritas, las que en esta última entidad también pueden persistir en el tiempo, dependiendo de la etiología y de la precocidad del inicio del tratamiento. La mucolipidosis tipo II es considerada como un trastorno autosómico recesivo con inicio en la infancia temprana, de curso lentamente progresiva y desenlace fatal desde la edad adulta temprana.19,20,21 Cathey yLeroy,reportaronensuestudiomuertes tempranas en pacientes con la misma enfermedad, con un pronóstico de vida incierto.22 El aumento y cronicidad en la secreción nasal y las infecciones recurrentes también son características clínicas de esta entidad, lo que se confirmó en el anterior reporte, las vías respiratorias son estrechasyestánsujetasaengrosamientos progresivos de la mucosa y la rigidez de todos los tejidos conectivos.23 Así mismo un gran porcentaje de los casos se puede desarrollar una enfermedad pulmonar intersticial progresiva.24
Debido a que los signos y síntomas de esta patología no están siempre presentes desde el nacimiento, a excepción de los grados más severos de la misma, su diagnóstico se realiza a través del tiempo y a medida que las manifestaciones clínicas son evidentes y que las pruebas de laboratorio e imágenes apoyen la impresión diagnostica.25 Estudios reportan muertes tempranas en pacientes con las mismas características del reporte, pero realmente el prostico de vida es incierto.26 Al paciente del caso se le realizaron pruebas de laboratorio debido a las alteraciones inespecíficas a nivel clínico inicialmente, donde se evidenció alteración de actividad enzimatica de beta-galactosidasa, Galactosa 6 sulfatasa, arilsulfatasa B, alfa L idurinidasa en suero y orina, niveles limítrofes superiores de esfingomielinasa acida e Iduronato sulfatasa, detectandose con mayor elevación en suero a la Arilsulfatasa A, por lo cual se remite a genética para realizar estudio panel NGS, en el cual se analizaron 4 genes: GNPTAB, GNPTG, MCOLN1 Y NEU1 (Figura 13). El reporte mostró la existencia de una mutación homocigota en el gen GNPTAB dando como diagnostico Mucolipidosis tipo II. Se inició tratamiento paliativo multidisciplinario por Medicina, Odontología, Fisioterapia, Oftalmología y Nutrición, para mejorar calidad de vida del paciente.
| Gen | Tipo de variante | Profundidad | Cambio en el ADN | Cigosidad | Cambio en proteína | dbSNP | Clinvar |
| GNPTAB | Frameshift | 192X | c.3503_3504deITC | Homocigota | p.Leu1168GInfs°5 | rs34002892 | Patogénica |
Figura 13: Genes con hallazgos de variantes de secuencia clasificadas como patogénicas según el análisis bioinformático.
De acuerdo a lo anterior, la ML II no presenta un tratamiento específico, que evite su curso deletéreo. El manejo está destinado a mejorar la calidad de vida del paciente a través de la prevención de la rigidez articular, del manejo adecuado del estado nutricional, que requiere habitualmente la instalación de una gastrostomía, y del tratamiento oportuno de infecciones respiratorias y urinarias de las eventuales convulsiones. No existe cura para la MLII y el tratamiento es paliativo. Los programas interactivos para estimular el desarrollo cognitivo, las terapias de ‘’bajo-impacto’’ (como la terapia acuática) y la terapia ocupacional y/o logopedia pueden resultar beneficiosos. Se puede valorar la gingivectomía en aquellos casos que cursan con dolor bucal grave e infecciones. En algunos pacientes se puede requerir apoyo respiratorio y ventilación asistida. Se debería realizar seguimientos periódicos a los bebés y niños pequeños (cada 3 meses) con el fin de monitorizar el funcionamiento cardiaco y pulmonar, y posteriormente cada año durante la primera infancia.
El estudio de las enfermedades de almacenamiento lisosomal es sumamente importante para comprender cuales son las consecuencias que puede causar un defecto genético que es expresado en el mal funcionamiento de un organelo, en este caso los lisosomas, ya que debido a su rara frecuencia en la cual se manifiesta estas enfermedades en la población no se tiene presente su comportamiento, debido a esto es difícil crear un tratamiento efectivo para dichas enfermedades. Se debe tener en cuenta que la calidad de vida de las personas que la padecen es de muy bajas expectativas y por esta razón es necesario ampliar el estudio en este campo y por lo general sólo reciben cuidados de naturaleza paliativa. Aplicando estudios con métodos de vanguardia en diversos campos para unificarlos, podrían hacer realidad la idea de un tratamiento efectivo, enfocado a la edición genética, de esta forma se podrá combatir de raíz una enfermedad de carácter autosómica recesiva como lo es la mucolipidosis y de esta forma tener certeza de obtener un resultado con una efectividad muy alta y tal vez definitiva a esta enfermedad.
Conflictos de interésLos autores declaran no tener ningún conflicto de intereses relacionado con el contenido del manuscrito.
Fuentes de financiaciónEste trabajo fue financiado por los recursos de los investigadores la Universidad del Sinú y la Universidad de Cartagena.