 Diana Elizabeth Fridman,2 Marcela Adriana Moya.3
Diana Elizabeth Fridman,2 Marcela Adriana Moya.3DOI:https://doi.org/10.47990/alop.v11i1.214
Ingrid Clarisa Guitelman,1 Diana Elizabeth Fridman,2 Marcela Adriana Moya.3
1 Jefe de Trabajos Prácticos, Cátedra Odontología,
Facultad de
Odontología, Integral Niños, Universidad de Buenos Aires, Buenos Aires,
Argentina.
2 Profesora adjunta, Cátedra Odontología, Facultad de
Odontología, Integral
Niños, Universidad de Buenos Aires, Buenos Aires, Argentina.
3 Jefe de Trabajos
Prácticos, Cátedra Odontología, Facultad de
Odontología, Integral Niños, Universidad de Buenos Aires, Buenos Aires, Argentina.
La Dentinogénesis Imperfecta es una anomalía dentaria determinada genéticamente y caracterizada clínica-mente por una apariencia ámbar opalescente de la dentina. Se presenta la resolución clínica, con seguimiento y control a 12 años de un paciente de 3 años de edad al momento de la consulta, con diagnóstico de Dentinogénesis Imperfecta tipo I asociada a Osteogénesis Imperfecta tipo IB. La identificación temprana de esta entidad y el tratamiento oportuno y multidisciplinario, contribuyen a mejorar el pronóstico de la misma.
Palabras clave: Dentinogénesis Imperfecta, Osteogénesis Imperfecta, multidisciplinario, niños.
Dentinogênese Imperfeita é uma anormalidade dentária geneticamente determinada, caracterizada clinicamente pela aparência opalescente e translúcida da dentina. Manejo clínico e seguimento de 12 anos são relatados, em um paciente de 3 anos com Dentinogênese Imperfeita tipo I associado à Osteogenesis Imperfecta tipo IB. O diagnóstico precoce.
Palavras chaves: Dentinogênese Imperfei-ta, Osteogenesis Imperfecta, multidiscipli-nar, criança.
Dentinogenesis Imperfecta is a geneti-cally determinated dental abnormality, characterized clinically by opalescent and translucent appearance of the den-tin. Clinical management and a 12 years follow up are reported, in a 3 years old patient with Dentinogenesis Imperfecta type I associated with Osteogenesis Im-perfecta type IB. The earlier diagnosis and the opportune and multidisciplinary treatment, led to improve the prognosis.
Key words: Dentinogénesis Imperfecta, Osteogénesis Imperfecta, multidisciplinary, children.
La Dentinogénesis Imperfecta (DI) es una anomalía dentaria hereditaria, caracterizada por un defecto en la formación de la matriz orgánica, durante la etapa de histodiferenciación en la odontogénesis, que trae como consecuencia una dentina anómala.1 Afecta generalmente a ambas denticiones, primaria y permanente.1
Clínicamente se caracteriza por la presencia de piezas dentarias con esmalte normal, ya que el trastorno es de origen mesodérmico, pero con dentina defectuosa; por esta razón el esmalte pierde soporte y tiende a fracturarse. La coloración translúcida propia de la enfermedad puede variar desde un azulado grisáceo o marrón opalescente al amarillo.2,3 Es frecuente observar piezas dentarias con un marcado desgaste incisal y oclusal, con pérdida de la estructura dentaria y disminución de la dimensión vertical.2
La histopatología muestra una cantidad reducida de túbulos dentinarios pero de mayor diámetro, que se presentan irregulares, desorientados y espaciados con áreas de matriz descalcificada.4 Si bien la dentina se forma continuamente, su matriz no está bien organizada, ni su grado de mineralización es el adecuado por lo cual hay un incremento del contenido orgánico y de agua y una disminución del componente inorgánico.
Radiográficamente las piezas dentarias presentan un signo patognomónico, en el que se evidencian coronas dentarias bulbosas con forma de campana, marcada constricción cervical, raíces cortas y cámaras pulpares que se obliteran aún en pacientes jóvenes.5 Como consecuencia del aumento del contenido orgánico y la reducción del inorgánico puede observarse falta de contraste.5
La DI se clasifica en 3 tipos:6
DI Tipo I: asociada a Osteogénesis Imperfecta (OI)
DI Tipo II: También conocida como dentina opalescente hereditaria, es la más frecuente y se presenta como una anomalía dentaria de estructura, determinada por un gen específico autosómico dominante no ligado al sexo.
DI Tipo III: menos frecuente y severa que la II, asociada a un grupo étnico de EE.UU.
La DI tipo II y III son causadas por mutaciones en el gen DSPP (4q12-21, sialofosfoproteína) que codifica a las proteínas principales (fosfoproteína dentinaria y dentina sialoproteína) implicadas en la formación de la dentina. 1,4,7
La DI tipo I es el fenotipo dentario registrado en pacientes con OI.1,8
La OI es un desorden genético hereditario, que afecta la integridad del tejido conectivo, caracterizado por la insuficiente o defectuosa formación del colágeno. Es el resultado de una mutación en el gen que codifica el colágeno tipo I (COL1A1 y COL1A2).8-10 Los tejidos en donde la principal matriz de proteínas es el colágeno tipo I (principalmente hueso, dentina y ligamentos) pueden estar afectados. En estos casos pueden encontrarse rigidez de los tejidos óseos, pérdida de la audición, DI, crecimiento deficiente, hiperlaxitud de articulaciones, y cualquier combinación de las mismas. La OI tiene múltiples características secundarias, incluyendo macrocefalia, escleróticas azules, defectos neurológicos (macrocefalia e invaginación basilar) y complicaciones cardiopulmonares.10
La clasificación original de Sillence de la OI incluía a los tipos I a IV11 sin embargo, en la última década se han identificado ocho tipos adicionales (V-XII) utilizando hallazgos histológicos y moleculares.12 En los tipos I a V, la OI tiene herencia autosómica dominante (AD). El descubrimiento de los tipos VI a XII reveló que las formas más raras de OI son autosómicas recesivas.10
Los pacientes con OI de tipo I tienen una forma distintivamente más leve de la enfermedad, que generalmente no es detectable al nacer. Tienden a presentar osteoporosis temprana, siendo frecuente la aparición de fracturas sobre todo en los años de preescolar que generalmente disminuyen drásticamente en los años posteriores a la pubertad. A menudo son unos pocos centímetros más bajos que los parientes del mismo sexo.10,13
La OI tipo I se divide en dos subtipos: subtipo A presenta dientes normales, mientras el subtipo B presenta DI.10
El objetivo de este trabajo es presentar el tratamiento integral y preventivo, realizado en un paciente de 3 años con DI Tipo I asociado a OI tipo IB, y su seguimiento durante 12 años restableciendo las funciones masticatorias, fonéticas, estéticas y la oclusión.
En el año 2007 se presentó a la Cátedra de Odontología Integral Niños (COIN) de la Facultad de Odontología de la Universidad de Buenos Aires, un niño de 3 años de edad, derivado del Hospital Juan P. Garrahan con diagnóstico de OI tipo I.
La historia médica reveló que la madre es portadora de OI tipo I con DI al igual que su hermana de 6 años. El diagnóstico en ellas, fue obtenido a partir de una fractura que había sufrido la hermana en el fémur a los 9 meses sumada a otra al año y medio de edad. Por esta razón los pediatras comenzaron a indagar acerca de situaciones de violencia doméstica, hipótesis que fue descartada luego que en el Hospital Juan P. Garrahan confirmaran el diagnóstico de OI.
El padre es sano. El diagnóstico del paciente se obtuvo después del nacimiento, dada la condición familiar y por tratarse de una enfermedad hereditaria. Desde temprana edad fue tratado en el Hospital Garrahan, presentando una forma leve de la enfermedad.
En la historia clínica de la COIN quedó registrado el consentimiento informado firmado por la madre del paciente, para la realización de procedimientos de diagnóstico, tratamiento, registro fotográfico y eventual publicación posterior.
En el examen extraoral se observaron escleróticas de tinte azulado y disminución de la altura con respecto al percentilo normal. (Figura 1) Ambos padres evidenciaban talla por debajo de los valores normales, más aún su madre, con antecedentes de OI; por lo que el niño además de su enfermedad de base la OI, presentaba baja talla por su condición hereditaria. En el peso, se hallaba en los valores promedio al igual que su estado físico general, situaciones que quedaron asentadas en el resumen de su historia clínica por los médicos especialistas que lo trataban en el Hospital Garrahan, del cual vino derivado. Por su patología de base el paciente realizaba controles periódicos sin tratamiento en el mencionado hospital y fue derivado a la COIN para su atención odontológica.
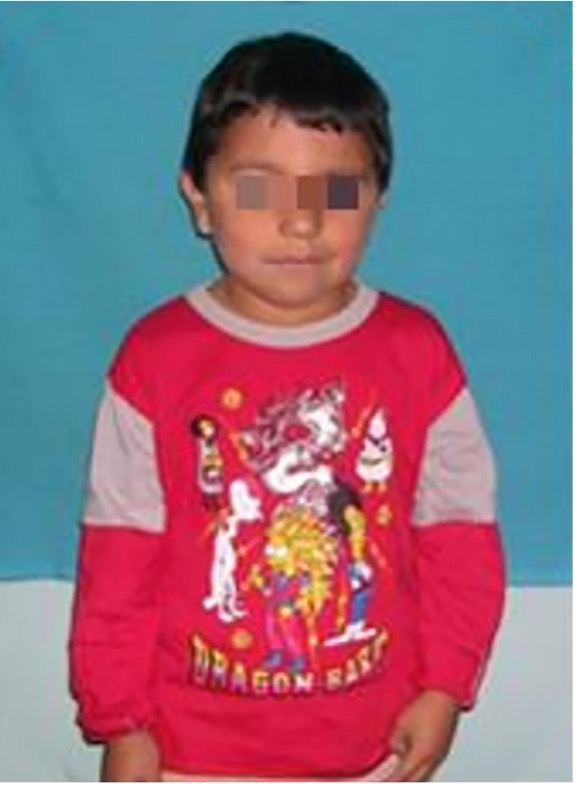

Figura 1: Examen extraoral del paciente. Escleróticas de tinte azulado
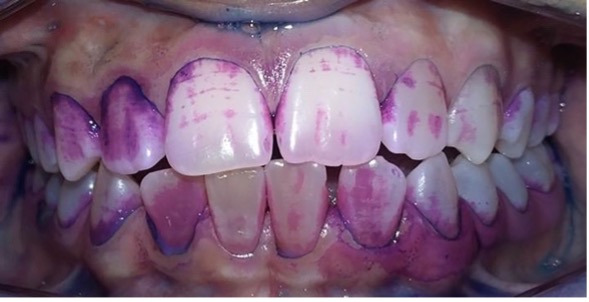
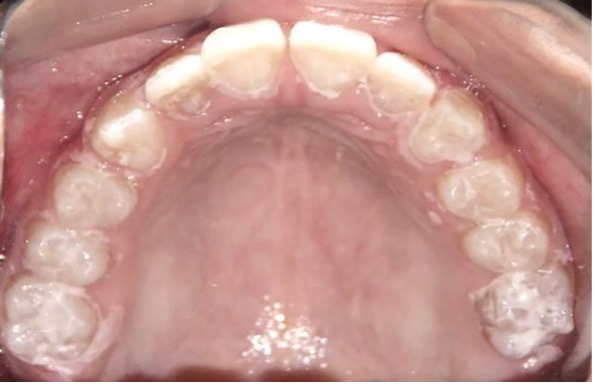
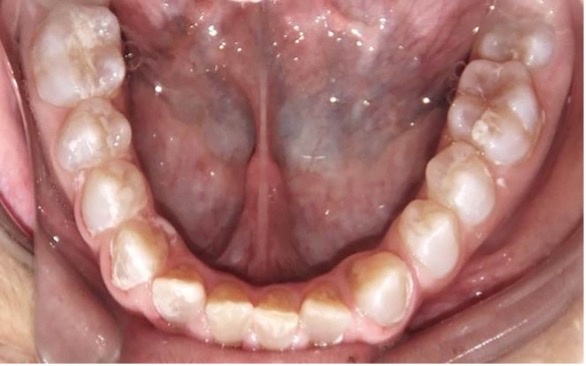
El examen clínico intraoral reveló alteración de la estructura y color de las piezas primarias, atrición y pérdida de la dimensión vertical (Figura 2). Radiográficamente se observó la presencia de coronas con marcada constricción cervical y raíces cortas (Figura 3). Presentaba alto riesgo cariogénico, por la presencia de lesiones de caries activas y dos restos radiculares, sumado a la anomalía estructural de la dentina (riesgo biológico específico) y bajo riesgo gingivoperiodontal. El riesgo socio-económico era alto por provenir de una familia con marcadas limitaciones económicas. Luego de la anamnesis, examen clínico y radiográfico, e interconsulta con el médico de cabecera se estableció el diagnóstico de DI Tipo I asociado a OI tipo I. Se planificó un tratamiento integral y preventivo con los objetivos de devolver forma y función, y proteger y evitar el desgaste de los tejidos conductas que pudieran interferir con el tratamiento.
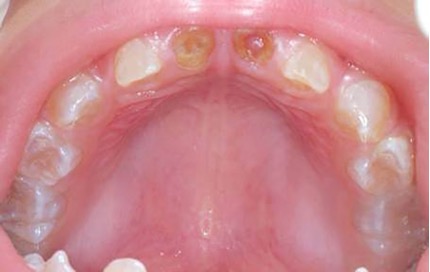
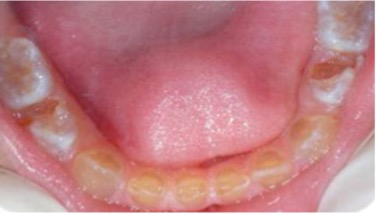
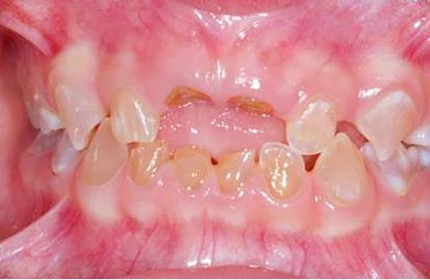
Figura 2: Examen clínico intraoral del maxilar superior e inferior y vista anterior. Año 2007
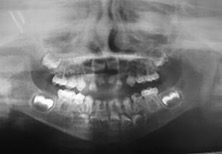
Figura 3: Radiografía panorámica año 2007
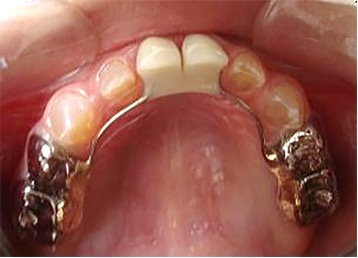
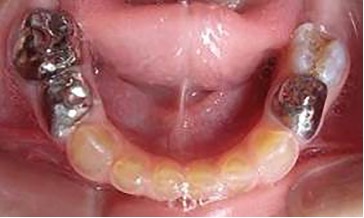
Figura 4: Postoperatorias clínicas del maxilar superior e inferior (2007)
En el año 2010, el mantenedor de espacio fijo superior se descementó, pero debido a la cercana erupción de los incisivos centrales superiores permanentes obser-vada radiográficamente (Figura 5), se decidió no recementarlo, confeccionando coronas de acero independientes en los 4 molares superiores primarios (5.4, 5.5, 6.4 y 6.5) y en el 7.5, previa restauración de las lesiones de caries con ionómero vítreo (Figura 6). Cuando se observó la erupción del 4.1 y 3.1, y de los 4 molares permanentes, se realizaron 3 aplicaciones de barniz de fluoruro de sodio al 5%, cumpliendo con los protocolos establecidos.
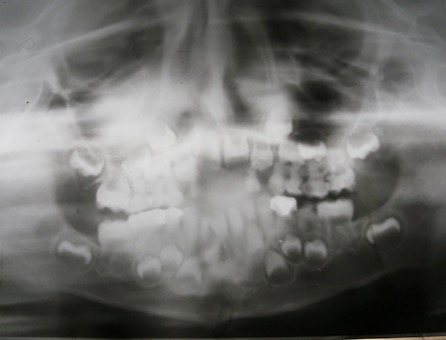
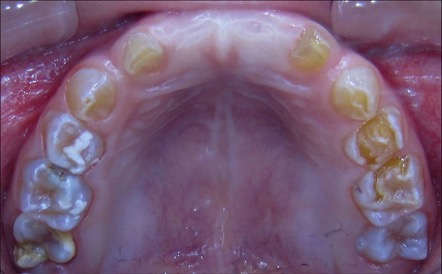
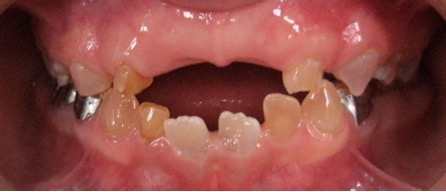
Figura 5: Examen clínico intraoral superior, sector anterior y radiografía panorámica año 2010.
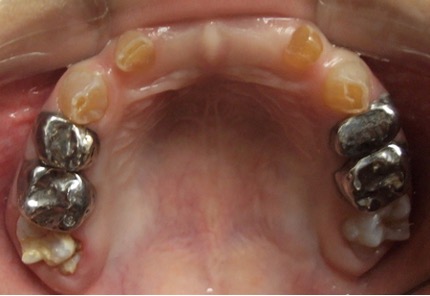
Figura 6: Postoperatorias clínicas maxilar superior e inferior año 2010.
En referencia al diagnóstico cráneofacial del paciente, el examen clínico extraoral en una vista frontal reveló: asimetría facial, con menor desarrollo del sector izquierdo, descenso de la ceja izquierda y el tercio inferior aumentado. Paciente compatible con biotipo dólico facial, obtusismo goníaco, rama ascendente corta, ligera mordida abierta, divergencia de bases, crecimiento rotacional posterior, ligera biprotrusión. En el examen intraoral se observó que la línea media se encontraba ligeramente desviada hacia la derecha. El pronóstico se consideró favorable en la medida que el paciente continuara con los controles periódicos (Figura 7). El paciente discontinuó el tratamiento durante los años 2011 y 2014. Cuando se reincorporó a la atención en la cátedra en el año 2015 se observó que en otro servicio se había realizado la exodoncia del 26 y que el 27 había erupcionado en esa posición (Figura 8) En este momento su riesgo cariogénico era alto por no haber acudido a los controles periódicos, presentar una nueva lesión de caries, más de 4 momentos de azúcar e historia pasada de caries. Los indicadores de presencia de placa e inflamación gingival revelaron un moderado riesgo gingivoperiodontal continuando alto su riesgo socio-económico. Se instauró un plan de tratamiento integral y preventivo haciendo énfasis en el cumplimiento del paciente a los controles integrales periódicos, monitoreando la higiene bucal y cumpliendo con los esquemas de intervención en los procesos de des-remineralización (aplicación de barniz de fluoruro de sodio al 5% (22600 ppm de Fˉ), dos veces por año) (Figura 9). Se restauró la caries en la pieza 3.6 con resina compuesta, previa base cavitaria con ionómero vítreo.
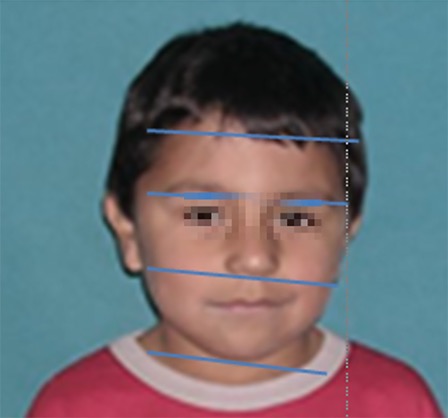
Figura 7: Estudio extraoral facial.
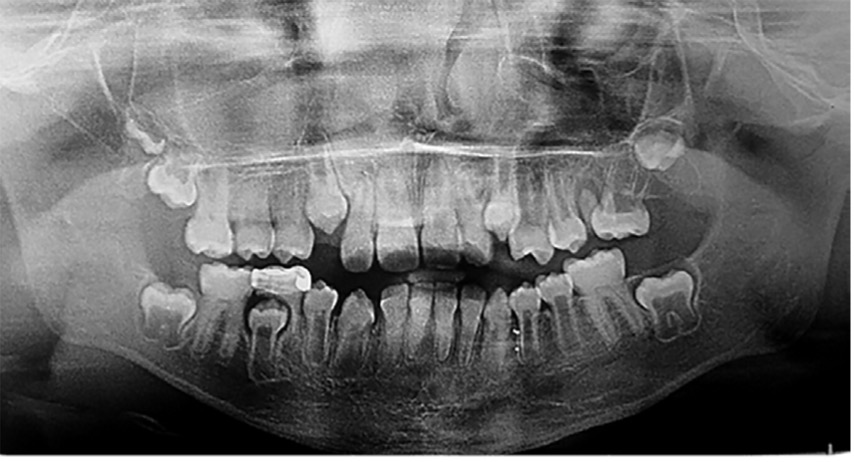
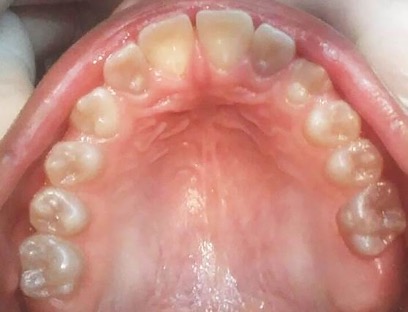
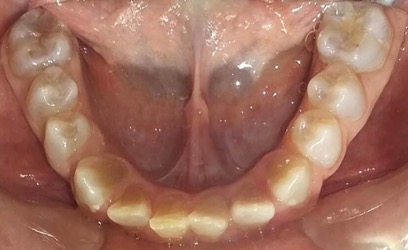
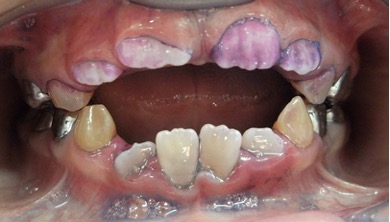
Figura 8: Radiografía panorámica (2015) y control clínico maxilar superior e inferior.
En la radiografía panorámica de control del año 2015 se observó la progresiva obliteración del conducto radicular en el grupo incisivo superior e inferior y en los primeros molares permanentes; presentado los premolares en erupción, un conducto radicular anormalmente amplio en toda su longitud (Figura 8). En la del año 2018 se visualizó la continua aposición de dentina a nivel radicular que obliteró en forma casi total los conductos de la mayoría de las piezas dentarias, a excepción de los segundos molares inferiores (Figura 10).
Control de placa bacteriana con revelador doble tono.
Aplicación de barniz de fluoruro de sodio.
Figura 9: Controles integrales periódicos
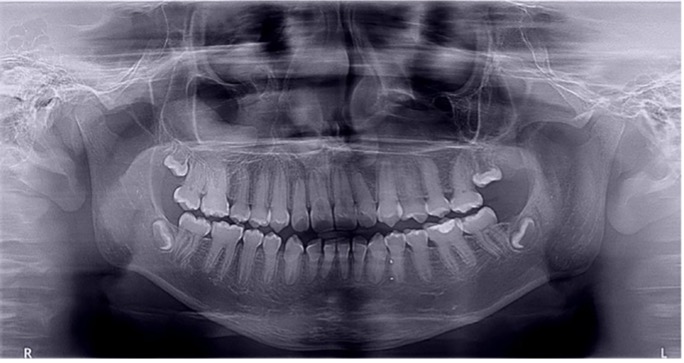
Figura 10: Rx panorámica 2018.
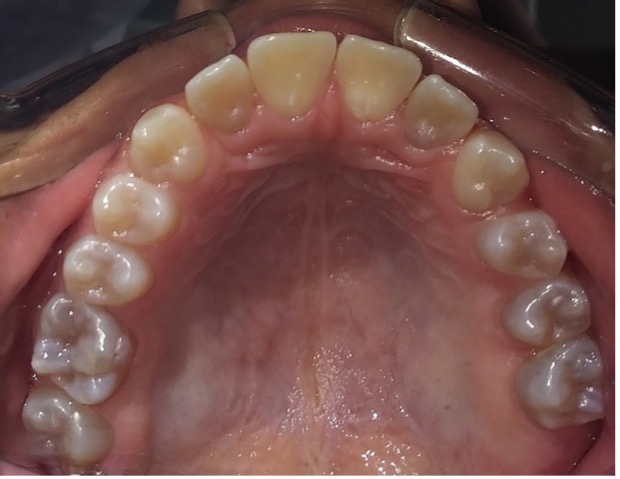
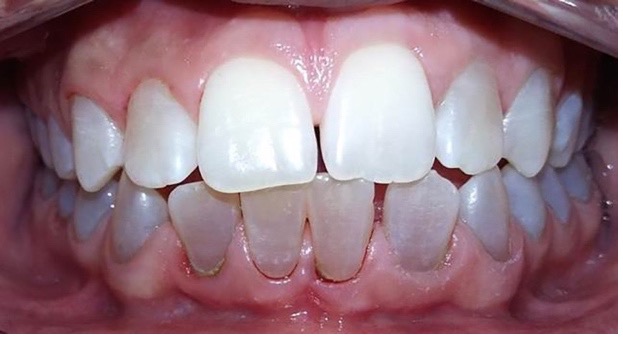
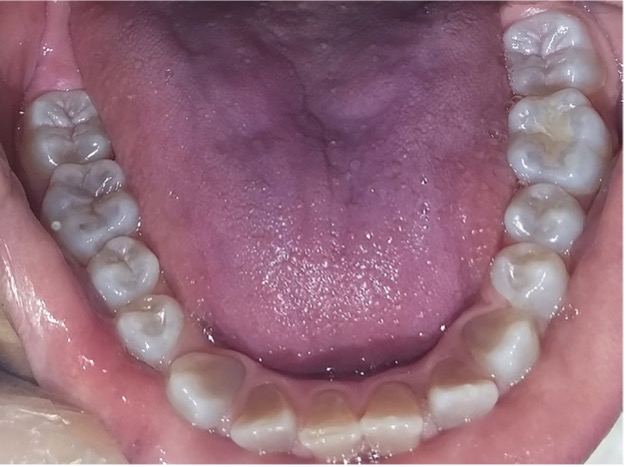
Figura 11: Controles clínicos 2018
En el último control realizado en el año 2018 (Figura 11) el paciente mantuvo el estado de salud logrado. El análisis cariogénico fue moderado (por su historia pasada de caries y su condición biológica específica) y el gingivoperiodontal bajo. El paciente continúa en atención en la COIN. Fue derivado en varias oportunidades al servicio de Ortodoncia de la Facultad de Odontología de la Universidad de Buenos Aires, pero hasta el momento no encaró ningún tratamiento propuesto.
El abordaje odontológico en pacientes con DI asociada a OI presenta numerosas variables dependiendo del grado de severidad de la expresión clínica. La identificación temprana y la instauración de medidas preventivas son esenciales, ya que contribuyen a minimizar las consecuencias funcionales y sociales propias de la enfermedad.1,14 El paciente recibió un tratamiento multidisciplinario, donde interactuaron odontopediatras, fonoaudiólogas, psicólogas, con inter-consultas con el médico de cabecera del Hospital Garrahan. Son numerosos los autores que coinciden en la necesidad del tratamiento interdisciplinario para un adecuado manejo de los pacientes con DI.1,3,5
Según la literatura si bien la DI tipo I involucra ambas denticiones, la dentición primaria se ve más seriamente comprometida2,4,9 situación que también se presentó en nuestro paciente. Debido a la gran pérdida de estructura dentaria y atrición de la dentición primaria, se optó por la colocación de coronas de acero en los molares primarios. Los superiores además formaron parte del mantenedor de espacio fijo para reponer las piezas 51 y 61. Las coronas de acero resultaron una medida eficaz que además de proteger a la estructura dentaria, compensaron la dimensión vertical disminuída.2, 3
Cuando la dentición permanente está implicada, los incisivos inferiores son los más comprometidos, presentando en estos casos una coloración intensa propia de la DI (O’Connell, Marini 1999, Majorana, Bardellini et al. 2010, Petersen, Wetzel 1998), como se observa en el paciente. Las piezas permanentes fueron monitoreadas desde su erupción, recibieron protocolos preventivos con barnices fluorados, pastas y enjuagatorios fluorados domiciliarios, conjuntamente con un adecuado control de higiene oral.15
Debido el alto porcentaje de abscesos periapicales, producto de la obliteración progresiva de los conductos radiculares, es fundamental el monitoreo radiográfico periódico en pacientes con DI.1 Los controles con radiografías panorámicas fueron realizados en el 2007, 2010, 2015 y 2018, sin registro de patologías pulpares periapicales.
Con respecto a su enfermedad de base que era la OI, al ser una forma leve, recibió controles sin medicación en el Hospital Garrahan.
Es imprescindible la identificación precoz de las anomalías dentarias y la instauración de un tratamiento temprano con abordaje multidisciplinario y con un alto componente preventivo. La protección del tejido dental con coronas de acero en la dentición primaria, evita el desgaste dentario y permite mantener la dimensión vertical.
A las Profesoras Dras. Ana María Biondi y Silvina Gabriela Cortese por la participación en la realización del trabajo.
Los autores no presentan conflictos de interés.